I hope you are both alright and in good health considering everything that is going on with the corona virus. I completely understand if you won't be able to answer my question because of health or working conditions.
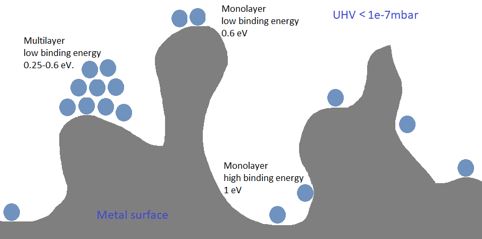
My question is about the binding energy of molecules to a surface. Binding energy of water on water is in the range of 0.25-0.6 eV. (sojourn times of microseconds) Binding energies for water on metal range from 0.6 to 1.05 eV or so (sojourn times from milliseconds to days).
There will be a range of binding energies (only interested in the monolayer) because of the shape of the surface on molecular level. I hope the picture will present some clarity of what I’m talking about.
Is it possible to insert a range of binding energies in the sojourn time area. For example in the same format you use for timesteps.
Start, increase, end --> for the binding energies (0.6,0.05,1.05) eV to J/mole conversion) 57890,4234,9000 J/mole in reality this is interpolated but 10 steps might be close enough.
Thanks for your concerns, we're doing fine, teleworking from home as CERN is in "safe mode", which means we're answering emails and forum posts faster than ever :)
The binding energy in Molfow is only an input variable to determine the mean sojourn time. Actual sojourn times are then generated following an exponential distribution (section 1.5 of this paper by C. Benvenutti: https://cds.cern.ch/record/454180/files/p43.pdf)
My point is that there is already a distribution of sojourn times.
What are you trying to achieve?
1) For a constant simulation, with multiple energies at the same time, you could either use the weighed average of the three binding energies, or do a very advanced hack: create multiple facets with tiny offsets ("layers", created by the Move Facet command towards their normal direction), each having a different binding energy. Then, you set the top layer's opacity to something lower than 1: in case of 0.6, for example, Molflow would "apply" the top layer with 60% probability and the bottom layer with 40%. (The particle wouldn't bounce between layers as one-sided facets are transparent from their back)
2) For a parameter sweep (series of constant simulations, each with a distinct binding energy), you have to set up and run each one manually, but an automatic parameter update is requested and planned.
3) For a time-dependent simulation, even if we added time-dependent binding energy, would it actually help? Molflow can only simulate processes up to a few seconds (1 minute at the very maximum, depending on the particle speed vs. the system's characteristic length). Do you really have a system where binding energy changes within seconds? This question also applies to your previous suggestion (time-dependent temperature).
 .
.