Hello again Chaoen:
"Did you publish any paper to explain your approach to obtain the steady state solution by counting the accumulated hits on the facets but completely forgetting the time sequence of individual virtual molecules."
The answer to this question, like many others, can be answered by looking at the best document on this, i.e. Marton's PhD thesis at EPFL Lausanne/CERN.
It can be downloaded (in case you didn't do it yet) here:
https://cds.cern.ch/record/2157666/files/CERN-THESIS-2016-047.pdf
Concerning your other question, how to implement the temperature gradient, it depends on the geometry of your model. If it is a tubular/cylindrical one then it is easy, it is sufficient to "cut" (virtually) the tube along the axis with perpendicular planes (using the "split facets" option in the "Facets" menu). You should select all facets on the side walls, of course.
You can split either using existing facets, using 3 vertices, or using the analytical formula for a plane in XYZ coordinates (which is what I do usually if the axis of my system is parallel to either one of the X, Y, or Z axis.... like I have done in this example (a 1 cm radius, 10 cm long tube):
https://www.dropbox.com/s/a2yzvrs9xsbf1vb/Reply_Chaoen_T_gradient.pdf?dl=0
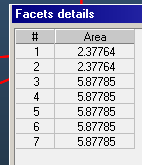
You repeat the procedure a suitable number of times (clearly, if you need a finely-tuned gradient with 100s of small temperature steps it will be very cumbersome), and then at the end you'll have to select each group of facets and assign to them, manually, a different temperature.
The same can be done quickly by selecting all facets on the side wall and then assigning to them a texture. If you select 1 as size of the texture (1 cm x 1 cm square texture elements), you should get a reasonably good accuracy... see dropbox document above.
Hope this solves your problem, otherwise feel free to write back!
Cheers,
Roberto